J. Geo. Chem. Soc., 2022, Vol. 2, Issue: 1, pp. 0 - 0
3,5-Bis(trifluoromethyl)phenyl ammonium triflate (BFPAT) as efficient organocatalysts for the combinatorial synthesis of xanthenes
Abstract. A green and efficient one-pot method has been developed for the synthesis of 1,8-dioxooctahydroxanthenes, 14-phenyl-14H-dibenzo[a,j]xanthene and tetrahydro benzoxanthene-11-ones using 3,5-bis (trifluoromethyl) phenyl ammonium triflate (BFPAT) as a highly efficient organocatalyst. The procedure offers several advantages, including a cleaner reaction profile, avoiding the use of typical toxic catalysts, moisture resistance, air tolerance, and low prices, which are the present method advantages.
Keywords: Organocatalyst, Green procedure, Xanthene, Multicomponent reactions, β-naphthol
Introduction
In the past decades, organocatalysis has attracted much attention as a robust methodology inorganic synthesis to construct new materials. In addition, small-molecule organocatalysts have provided opportunities to develop alternative and, in many cases, more practical synthetic approaches toward the construction of carbon-carbon and carbon-heteroatom bonds 1-4. Over the past few years, organocatalytic reactions affording achiral compounds have received considerable attention as an inexpensive, green, readily available agent affording the corresponding products with high selectivity in excellent yields [5].
effectivelyrepels
In recent years, aryl ammonium triflate has proved useful as a mild and efficient catalyst in organic synthesis [6-11]. The unique properties such as being readily affordable, water stability, recyclability, operational simplicity, strong tolerance to oxygen, and nitrogen-containing substrates and functional groups make them more advantageous than the conventional catalysts. As part of our continued interest in developing new reagents or systems for developing more efficient and environmentally benign methodologies, [12-15]we present herein an efficient method for the synthesis of 1,8-dioxooctahydroxanthene, 14-phenyl-14H-dibenzo[a,j]xanthene, and tetrahydro benzo xanthene-11-one derivatives utilizing 3,5-bis (trifluoromethyl) phenyl ammonium triflate (BFPAT), a new effective organocatalyst. Xanthenes are a class of important oxa-heterocycles possessing diverse biological activities, such as antiviral [16], anti-inflammatory [17], antibacterial activities [18], antiplasmodial [19], as well as their use as antagonists for drug-resistant leukemia lines [20]. Some xanthenes have also been used as dyes and fluorescent materials [21, 22]. Furthermore, xanthene moieties are essential structural fragments of a large number of pharmacologically active compounds, like RO67-4853(IC50 BHK cell 114.4), Rhodamine, and Amsacrine (IC50HCT-8 = 105 and L1210 = 32) [23, 24] (Figure 1).
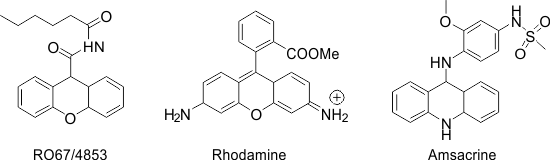
Figure 1: Bioactive compounds incorporating the xanthenes ring
Considering the above reports, there has been continuous interest in developing facile synthetic protocols for constructing xanthene derivatives. Over the past decade, many synthetic methods have been reported to prepare xanthene and its analogs [25, 26-35]. Recently, Chaturbhuj et al. [36] reported sulfated polyboratecatalyzed reaction between β-naphthol, aldehydes, and dimedone to prepare xanthene derivatives in a one-pot synthesis. Furthermore, Pitchumani et al. [37] developed an efficient procedure for synthesizing benzoxanthenone derivatives via the one-pot three-component condensation of naphthols, aldehydes, and cyclic 1,3-dicarbonylcompoundsusing HY zeolites as a catalyst. However, most of them suffer from the drawbacks such as strongly acidic conditions, expensive moisture-sensitive catalysts, tedious workup conditions, low yields, undesirable metal catalysts, and harsh reaction conditions.
Furthermore, many of them are not eco-friendly. In addition, most of the described methodologies result in large amounts of byproducts leading to a poor overall atom economy. Therefore, exploring a more general, efficient, and environmentally benign protocol for developing new, greener synthetic protocol is highly desirable. Herein, we report an efficient and facile synthesis of 1,8-dioxooctahydroxanthenes, 14-phenyl-14H-dibenzo[a,j]xanthene, and tetrahydrobenzoxanthene-11-ones using BFPAT as a novel organocatalyst (Scheme 1).
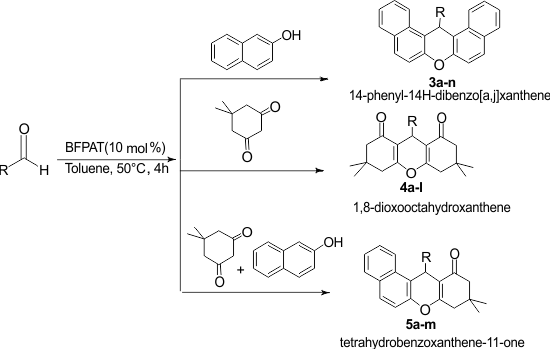
Scheme 1. Synthesis of xanthenes using BFPAT organocatalyst
- Results and discussion
In preliminary experiments, the model reaction of β-naphthol (2.0 mmol) and benzaldehyde (1.0 mmol) catalyzed by BFPAT was conducted to screen the optimal reaction conditions. The results were listed in Table 1. It is important to note that the reaction failed to give the desired product without a catalyst, even after a long reaction time (24 h; Table 1, entry 1). We next evaluated the effect of temperature, the reaction time, and the amount of catalyst on the yield of the product, and the results are summarized in Table 1.
Table 1. Effect of different BFPAT and solvent on the formation of 4
|
Entry |
BFPAT amount (mg) |
BFPAT amount (mg) |
Time (h)/yield |
|
1 |
0 |
rt/toluene |
24/0 |
|
2 |
5 |
rt/toluene |
24/20 |
|
3 |
5 |
50° C/toluene |
5/40 |
|
4 |
10 |
50° C/toluene |
4/90 |
|
5 |
10 |
rt/CH2Cl2 |
5/20 |
|
6 |
10 |
rt/diethylether |
5/10 |
|
7 |
10 |
reflux/H2O |
5/50 |
|
8 |
10 |
reflux/ethanol |
5/65 |
|
9 |
15 |
50° C/tolune |
5/91 |
The effect of various solvents on time and yield were also investigated (e.g., CH2Cl2, toluene, ethanol, diethyl ether, and H2O) and (Table 1, entries 2–9). Toluene provided the highest yield at 50°C after 4 h (Table 1, entry 4). Due to the low solubility of β-naphthol in toluene at room temperature, the reaction does not proceed—while increasing the temperature to 50 °C caused increased product yield in a shorter reaction time. Next, we examined the effect of the catalyst loading on time and yield using the same model reaction. While increasing the optimal amount of catalyst to 10mol %, the products of 3a were afforded in 90% yield, respectively (entry 4), which showed the critical role of catalyst concentration in the reaction. No improvement in yield was observed when the reaction was carried out at a reduced temperature or decreasing the amount of catalyst (Table 1, entries 2, 3). To explore the applicability of our reaction, we employed a variety of substituted aliphatic and aromatic aldehydes with dimedone or β-naphthol for the synthesis of a wide variety of substituted14-substituted-14H-dibenzo[a,j]xanthenes, and results are summarized in Table 2 and 3.
A wide range of structurally varied aldehyde reacted smoothly and quickly to give the corresponding 14-substituted-14H-dibenzo[a,j]xanthenes in high yield and purity, as listed in Table 2.
Table 2: Synthesis of 14-phenyl-14H-dibenzo[a,j]xanthenein the presence of BFPAT
|
Entry |
Carbonyl Compound |
Product |
Yield, % |
mp, °C |
|
1 |
3a |
90 |
184-185 |
|
|
2 |
3b |
91 |
294-296 |
|
|
3 |
3c |
90 |
313-314 |
|
|
4 |
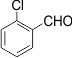 |
3d |
80 |
216-218 |
|
5 |
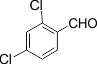 |
3e |
85 |
267-268 |
|
6 |
3f |
90 |
236-237 |
|
|
7 |
3g |
85 |
213-215 |
|
|
8 |
3h |
92 |
296-297 |
|
|
9 |
3k |
85 |
138-140 |
|
|
10 |
3l |
85 |
222-224 |
|
|
11 |
3m |
83 |
202-204 |
|
|
12 |
3n |
88 |
172-174 |
Table 3: Synthesis of 1,8-dioxooctahydroxanthenesin the presence of BFPAT
|
Entry |
Carbonyl Compound |
Product |
Yield, % |
mp, °C |
|
1 |
|
4a |
90 |
184-185 |
|
2 |
|
4b |
91 |
294-296 |
|
3 |
|
4c |
85 |
313-314 |
|
4 |
|
4d |
88 |
296-297 |
|
5 |
|
4e |
90 |
236-237 |
|
6 |
|
4f |
93 |
267-268 |
|
7 |
|
4g |
92 |
213-215 |
|
8 |
|
4h |
90 |
185-187 |
|
9 |
|
4k |
92 |
296-297 |
|
10 |
|
4l |
90 |
222-224 |
The electronic properties of the substituents on the aromatic ring system were shown to have little influence on the efficiency of this reaction. For example, Aromatic aldehydes bearing an electron-neutral (H) or electron-donating (e.g., 4-Me, 4-OMe, and 4- OH) group reacted smoothly to give the corresponding products in good to excellent yields (83−85%; 3l−3m). The experimental procedure is very efficient, convenient, and rapid. In addition, it can tolerate a variety of other functional groups, such as alkyl methoxy, nitro, and halides, under these reaction conditions. Satisfactory high yields were also observed for substrates bearing an electron-withdrawing group (e.g., 4-Cl, 3-NO2, 4-NO2, and4-Br) on the benzene ring (85−92%; 3b-3k). Notably, electron-serially hindered (2-Cl and 2,4-di-Cl) substrates reacted well to afford the expected products in moderate to good yields (80−85%; 3d−3e).
Encouraged by this success, we attempted to synthesize tetrahydrobenzoxanthene-11-ones 5 from β-naphthol aldehydes and dimedone under similar conditions (Table 4). Again, good yields were obtained for all cases; all the aldehydes gave the corresponding 12-aryl-8,9,10,12-tetrahydrobenzo[a]xanthen-11-ones as the major products.
Table 4: Synthesis of tetrahydrobenzoxanthene-11-ones in the presence of BFPAT
|
Entry |
Carbonyl Compound |
Product |
Yield, % |
mp, °C |
|
1 |
5a |
90 |
150-152 |
|
|
2 |
5b |
88 |
177-179 |
|
|
3 |
5c |
85 |
205-206 |
|
|
4 |
5d |
80 |
239-240 |
|
|
5 |
5e |
92 |
180-182 |
|
|
6 |
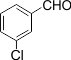 |
5f |
85 |
174-176 |
|
7 |
5g |
88 |
178-179 |
|
|
8 |
5h |
90 |
180-182 |
|
|
9 |
5k |
80 |
168-170 |
|
|
10 |
5l |
95 |
186-188 |
|
|
11 |
5m |
90 |
184-185 |
The structure of the products was characterized by their IR spectral data and a comparison of their melting points with those of authentic samples [36]. A plausible mechanism of catalyst working is depicted in Scheme 2.
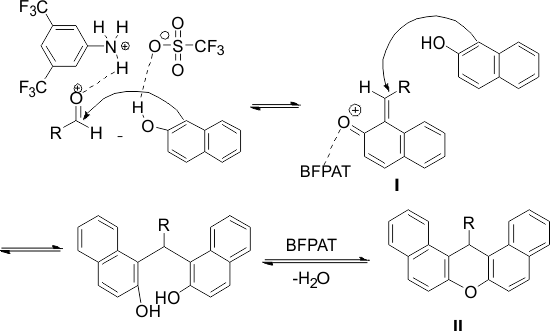
Scheme 2. Probable mechanism for BFPAT-catalyzed Synthesis of 14-phenyl-14H-dibenzo[a,j]xanthenes
In this process, BFPAT can increase the electrophilic character of the electrophiles by its inherent Brønsted acidity, which makes it capable of bonding with the carbonyl oxygen. Moreover, the highly hydrophobic wall of the moiety of BFPAT effectively repels H2O produced by the condensation. The superiority of BFPAT to similar catalysts such as pentafluorophenyl ammonium triflate (PFPAT) [6]and diphenyl ammonium triflate (DPAT) [38] is ascribed to the lower basicity of the (CF3)2C6H3NH2 counter amine compared to C6F5NH2 and Ph2NH. Therefore, it was supposed that product 3 might be formed via the initial formation of intermediate I. First, the aldehyde is activated by BFPAT, followed by the carbonyl carbon is attacked by the nucleophilic β-naphthol (activated by BFPAT) to form intermediate I. Then, the subsequent addition of these fragments to another mole of β-naphthol gives the acyclic adducts intermediate, which undergoes intramolecular dehydrative cyclization to afford the xanthene derivatives (II). Therefore, it may be assumed that the water exclusion of BFPAT may favor both cyclization and intermediate I formation. In addition, (CF3)2C6H3NH2 easily separated from the reaction mixture after work-up with distillation under reduced pressure ((CF3)2C6H3NH2: 85 °C/15 mmHg (lit.)).
In summary, we have demonstrated an extremely efficient and green process for synthesizing 1,8-dioxooctahydroxanthene, 14-phenyl-14H-dibenzo[a,j]xanthene, and tetrahydro benzoxanthene-11-one derivatives employing BFPAT as a novel organocatalyst in a one-pot fashion. The significant features of this protocol are: (1) using BFPAT as a readily available, inexpensive, and efficient organocatalyst, (2) simple reaction condition, (3) green and cost-effective catalyst, and (4) easy purification, and excellent yields. In addition, we wish to state that this method involves an environmentally friendly procedure and is the first procedure for synthesizing xanthene derivatives.
3. Experimental
Preparation of 3,5-bis(trifluoromethyl) phenylammoniumtriflate (BFPAT): CF3SO3H (7.50 g, 50 mmol) was added to a stirred solution of 3,5-bis(trifluoromethyl)aniline (11.45 g, 50 mmol) in toluene(30.0 ml) at 0–5 °C, and the mixture was stirred at the same temperature for 30 min. Evaporation of the solvent gave the crude product, washed with dry ether to give a pure BFPAT (18.4 g, 90%) as colorless crystals. 1H NMR (400 MHz, DMSO-d6): δ: 7.03 (s, 1H), 7.14(s, 2H); IR (KBr) 3416, 2965, 1532, 1250, 1179 cm-1.
General procedure for the preparation of 14-aryl (alkyl)-14H-dibenzo[a,j]xanthenes and 1,8-dioxooctahydroxanthenes
A toluene solution (3 ml) of aldehyde (1 mmol) andβ-naphthol (2 mmol) or 5,5-dimethyl-1,3-cyclohexanedione (2 mmol)was mixed with BFPAT (10 mol%), and the resulting mixture was stirred at 50°C for an appropriate time. Upon completion of the reaction, as indicated by TLC, the organic phase was washed with 1 mol/L aqueous NaOH solution (1 ml). The organic phase was then collected. The solvent was removed in vacuo to give the crude product, which was purified by recrystallization from hot ethanol to afford the pure product.
General procedure for the preparation of 12-aryl-8,9,10,12-tetrahydrobenzo[a]xanthen-11-one derivative
A mixture of aldehyde(1 mmol), β-naphthol (2 mmol), and 5,5-dimethyl cyclohexane-1,3-dione dissolved in 3mL toluene, and BFPAT (10 mol %) was stirred for 4 h at 50 °C. TLC monitored the reaction. After being cooled to room temperature, the reaction mixture was poured onto crushed ice and stirred for 5–10 min. The crystalline product was collected by filtration under suction (water aspirator), washed with ice-cold water (40 ml), and then recrystallized from hot ethanol to afford pure products. The filtrate was concentrated under reduced pressure and then recrystallized from hot hexane to recover the BFPAT for subsequent use.
Spectroscopic data for selected examples follow:
Compound 3a: Colorless solid; mp 185–187 °C. (KBr, cm–1): υ 3071, 1585, 1444, 1250, 1080 cm_1. 1H NMR (CDCl3, 400 MHz):δ 6.45 (s, 1H), 6.96 (t, J = 7.2 Hz, 1H), 7.12 (t, J = 7.2 Hz, 2H), 7.37–7.57 (m, 8H), 7.75–7.83 (m, 4H), 8.34 (d, J = 8.5 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 147.9, 145.4, 130.8, 130.6, 128.9, 128.5, 128.3, 127.9, 126.8, 126.1, 124.4, 123.4, 117.6, 117.3, 36.5
Compound 3b: Yellow solid; mp 291–292°C. IR (KBr, cm–1): 3061, 2919, 1623, 1590, 1515, 1450. 1H NMR(400 MHz, CDCl3): δ6.76 (s, 1H), 7.17 (d, J= 6.8 Hz, 2H), 7.44–7.63 (m, 10H), 8.91 (d, J = 7.8 Hz, 2H),8.66 (d, J = 7.6 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 148.5, 144.7, 131.3, 131.2, 130.1, 129.6, 129.2, 128.8, 128.2, 125.1, 123.8, 123.5, 118.1, 117.1, 36.3.
Compound 3c: Yellow solid; mp 312–313 °C. (KBr, cm–1): υ 2926, 1589, 1516, 1335. 1H NMR (CDCl3, 400 MHz) δ 6.59 (s, 1H), 7.03 (t, J = 7.8 Hz, 2H), 7.40–7.56 (m, 6H), 7.66–7.72(m, 4H), 8.30–8.46 (m, 4H).13C NMR (100 MHz, CDCl3): δ151.9, 148.7, 131.0, 129.5, 129.0, 128.9, 127.1, 124.5, 123.8, 122.0, 118.0, 115.7,
37.8
Compound 3d: White solid; mp 216–218 °C. (KBr, cm–1): υ 3055, 2995, 1595, 1510, 1459, 1240, 961. 1H NMR (CDCl3,400 MHz):δ 6.80 (s, 1H), 6.87–7.13 (m, 2H), 7.42–7.67 (m, 8H),7.79–7.86 (m, 4H), 8.73 (d, J = 8.5 Hz, 2H).13CNMR (CDCl3, 400 MHz):δ148.8, 143.3, 131.8, 131.7, 130.8, 130.1, 129.6, 129.1, 128.5, 127.8, 127.6, 126.7, 124.3, 123.2, 34.5.
Compound 3k: Pink solid; mp 138–140 °C. (KBr, cm–1):υ 3403,1591, 1512, 1401, 1250, 1242. 1H NMR (CDCl3,400 MHz):δ 4.98 (s, 1H), 6.42 (s, 1H), 6.58 (d, J = 8.4 Hz, 2H),7.38 (t, J = 10.8 Hz, 2H), 7.43 (d, J = 7.5 Hz, 2H), 7.48 (d, J = 8.9, 2H),7.58 (t, J = 7.4 Hz, 2H), 7.79 (d, J = 5.1 Hz, 2H), 7.83 (d, J = 8.1 Hz,2H), 8.39 (d, J = 8.4 Hz, 2H).13C NMR (100 MHz, CDCl3): δ 154.1, 149.2, 137.8, 131.5, 131.2, 129.9, 129.3, 129.1, 127.2, 124.6, 123.4, 118.4,117.5, 115.5, 37.5.
Compound 3m: White solid; mp 203–206 °C.(KBr, cm–1):υ 3036,2911, 1610,1580, 1240. 1H NMR (CDCl3, 500 MHz):δ 3.72 (s, 3H), 6.48 (s, 1H), 6.75 (d, J = 7.5 Hz, 2H), 7.39 (t, J = 7.2 Hz,2H), 7.53–7.89 (m, 10H), 8.41 (d, J = 8.5 Hz, 2H).13C NMR (100 MHz, CDCl3): δ 148.6, 142.1, 135.8, 131.5, 131.1,129.1, 128.5, 128.1, 126.5, 124.2, 123.1, 118.0, 117.5, 37.5, 20.9
Compound 4a: White solid; mp 204–205 °C. IR (KBr, cm–1): υ2954, 1664, 1364, 1199. 1H NMR (400 MHz, CDCl3): δ 0.90 (s, 6H), 1.04 (s, 6H), 2.09 (d, J = 16.2 Hz, 2H), 2.27 (d, J = 16.2 Hz, 2H), 2.53 (d, J = 17.2 Hz, 2H), 2.58 (d, J = 17.7 Hz, 2H), 4.53 (s, 1H), 7.10 (t, J = 7.0 Hz, 1H), 7.18 (d, J = 7.0 Hz, 2H), 7.22 (t, J = 7.20 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 196.8, 162.7, 144.5, 128.8, 128.4, 126.8, 116.1, 51.2, 41.3, 32.6, 32.3, 29.6, 27.7.
Compound 5a: White solid; mp 150–152 °C; IR (KBr, cm-1): υ 3,055, 2,950, 2,879, 1,650, 1,376. 1HNMR (400 MHz, CDCl3): δ 8.01 (d, J = 8.1 Hz, 1H), 7.73–7.78 (m,2H),7.05–7.40 (m, 8H), 5.73 (s, 1H), 2.61 (s, 2H), 2.33 (d, J = 16.1 Hz, 1H),2.25 (d, J = 16.1 Hz, 1H), 1.12 (s, 3H), 0.93 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 196.6, 164.1, 147.5, 144.3, 132.1, 131.3, 128.8, 128.4, 128.2, 128.0, 126.6,126.1, 124.8, 123.5, 117.3, 117.1, 114.1, 50.5, 41.5, 34.7, 32.3, 29.6, 27.2.
Compound 5c: White solid; mp 205–206 °C; IR (KBr, cm-1): υ 3,056, 2,948, 1,642, 1,224, 1,170. 1HNMR (400 MHz, CDCl3): δ 0.98 (s,3H), 1.13 (s, 3H),2.27 (d, J = 16.2 Hz, 1H),2.35 (d, J = 16.2 Hz, 1H),2.56 (s, 2H),3.68 (s, 3H),5.64 (s, 1H),6.71 (d, J = 8.2 Hz, 2H),7.21–7.42 (m, 5H),7.71–7.76 (m, 2H),8.01 (d, J = 8.1 Hz, 1H). 13CNMR (100 MHz, CDCl3): δ 196.5, 164.1, 155.6, 146.3, 136.2,133.6, 130.6,130.1, 128.5, 127.2, 126.3, 125.1, 123.6, 122.2, 117.1, 116.8, 115.4, 113.2, 112.7,53.4, 49.5, 40.2, 32.5, 31.1, 28.4, 26.2.
Compound 5e: White solid; mp 180–182 °C; IR (KBr, cm-1):υ 3070, 2935, 1640, 1368. 1H NMR(400 MHz, CDCl3): δ 0.95 (s, 3H), 1.12 (s, 3H),2.25 (d, J = 16.1 Hz,1H),2.33 (d, J = 16.1 Hz, 1H),2.56 (s, 2H),5.65 (s, 1H),7.10–7.42(m, 7H),7.75–7.80 (m, 2H),7.89 (d, J = 8.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 196.7, 164.2,152.0, 147.1, 146.2, 132.1, 130.2, 129.1, 129.2, 128.3, 127.3, 125.1, 123.1, 123.0,116.2, 115.3, 113.1, 50.2, 41.5, 34.7, 32.1, 29.3, 27.2.
Compound 5f: White solid; mp 175–177 °C; IR (KBr, cm-1): υ 3050, 2927, 1647, 1365. 1H NMR(400 MHz, CDCl3): δ 0.98 (s, 3H),1.11 (s, 3H), 2.13 (d, J = 16.2 Hz, 1H), 2.30 (d, J = 16.2 Hz, 1H), 2.60 (s,2H), 5.60 (s, 1H), 7.11 (s, 1H), 7.19 (d, J = 4.4 Hz, 2H), 7.36 (s, 1H), 7.40–7.50(m, 3H), 7.88–8.01 (m, 2H), 8.03 (d, J = 8.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 195.5, 164.1, 148.1, 148.0, 134.5, 132.2,131.3, 130.5, 130.3, 129.2, 128.5, 128.1, 127.4, 127.1, 125.9, 124.2, 117.5, 117.2,113.4, 51.5, 42.1, 35.5, 33.7, 29.8, 26.8.
Compound 5g: White solid; mp 178–180 °C; IR(KBr, cm-1):υ 3070, 2929, 1645, 1369, 1230. 1H NMR (400 MHz, CDCl3): δ0.98 (s, 3H), 1.12 (s, 3H),2.24 (d, J = 16.0 Hz, 1H),2.34 (d,J = 16.0 Hz, 1H),2.60 (s, 2H),5.88 (s, 1H),6.95–7.07 (m, 2H),7.27–7.49 (m, 5H),7.71–7.75 (m,2H),8.22 (d, J = 8.3 Hz, 1H); 13C NMR(100 MHz, CDCl3): δ 196.4, 164.2, 147.8, 142.5, 133.1, 131.3,123.1, 131.5, 131.2, 129.3, 129.0, 128.1, 127.2, 127.1, 126.5, 124.9, 122.9, 117.1, 117.0, 14.1, 51.1, 41.4, 32.5, 32.2, 29.4, 27.2.
Compound 5h: White solid; mp 177–180 °C; IR(KBr, cm-1): υ3078, 2933, 2911, 1645, 1595,1510. 1H NMR (400 MHz, CDCl3): δ 8.03 (d, J = 8.4 Hz, 2H),7.78–7.80 (m, 3H), 7.32–7.51 (m, 5H), 5.80 (s, 1H), 2.60 (s, 2H), 2.35 (d,J = 16.3 Hz, 1H), 2.24 (d, J = 16.3 Hz, 1H), 1.11 (s, 3H), 0.93 (s, 3H). 13C NMR(100 MHz, CDCl3): δ 196.2, 165.1, 51.6, 147.5, 145.9, 131.4, 130.9, 129.6,129.3, 128.4, 127.1, 125.2, 123.2, 122.9, 117.3, 115.9, 113.0, 50.7, 41.2, 34.6, 32.1,29.3, 27.1.
Compound 5k: White solid; mp 168–170 °C; IR (KBr, cm-1): υ3073, 2950, 1648, 1527, 1370. 1H NMR (400 MHz, CDCl3): δ 8.09 (s, 1H), 7.79–7.94 (m, 5H),7.35–7.46 (m, 4H), 5.80 (s, 1H), 2.60 (s, 2H), 2.36 (d, J = 16.2 Hz, 1H), 2.27 (d,J = 16.2 Hz, 1H), 1.14 (s, 3H), 0.97 (s, 3H). 13C NMR (100 MHz, CDCl3):δ 196.4, 164.2, 148.5, 147.7, 146.5, 134.5, 132.1, 130.6, 129.6, 129.1, 128.4,127.2, 125.1, 123.2, 123.0, 121.2, 117.2, 115.5, 113.2, 51.2, 42.4, 34.5, 32.1, 29.1,27.2.
Compound 5l: White solid; mp 186–187 °C; IR (KBr, cm-1): υ 3072, 2936, 1645, 1372.1H NMR(400 MHz, CDCl3): δ 0.96 (s, 3H), 1.13 (s, 3H),2.24 (d, J = 16.3 Hz, 1H),2.31 (d,J = 16.3 Hz, 1H),2.56 (s, 2H),5.66 (s, 1H),7.26–7.33 (m, 3H),7.38–7.45 (m,2H),7.76–7.80 (m, 2H),7.88 (d, J = 8.3 Hz, 1H). 13C NMR(100 MHz, CDCl3): δ 196.8, 164.1, 147.5, 143.8, 131.5, 131.3, 131.2, 130.1, 129.3,128.5, 127.2, 125.0, 123.4, 120.2, 117.1, 116.8, 113.7, 50.8, 41.4, 34.3, 32.5, 29.2, 27.1.
Compound 5m: White solid, m.p. 184–185 °C; IR (KBr, cm-1): υ3034, 2952, 2880, 1654, 1620. 1H NMR (400 MHz, CDCl3): δ0.97 (s, 3H), 1.13 (s, 3H),2.31 (d, J = 16.4 Hz, 1H),2.24 (d, J = 16.4 Hz, 1H),2.55 (s, 2H),5.72 (s, 1H),6.86 (t, J = 8.6 Hz, 2H),7.30–7.45 (m, 5H),7.76 (t,J = 8.2 Hz, 2H),7.98 (d, J = 8.4 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 196.8, 163.6, 148.1, 141.6, 132.5, 131.7, 129.8, 129.2, 128.5, 127.2, 125.2, 123.5, 117.5, 117.2, 115.2, 114.9, 114.2, 51.2, 41.5, 34.3, 32.1, 29.2, 27.1.
Acknowledgments
The authors thank the Research Committee of Islamic Azad University (Ahvaz&Ayatollah Amoli Branch) and the University of Georgia (Tbilisi) for financial support of this work.
References:
- Dalko, P.I. and L. Moisan, In the golden age of organocatalysis. Angewandte Chemie International Edition, 2004. 43(39): p. 5138-5175.
- Doyle, A.G. and E.N. Jacobsen, Small-molecule H-bond donors in asymmetric catalysis. Chemical Reviews, 2007. 107(12): p. 5713-5743.
- Peng, F. and Z. Shao, Advances in asymmetric organocatalytic reactions catalyzed by chiral primary amines. Journal of Molecular Catalysis A: Chemical, 2008. 285(1-2): p. 1-13.
- Dondoni, A. and A. Massi, Asymmetric organocatalysis: from infancy to adolescence. Angewandte Chemie International Edition, 2008. 47(25): p. 4638-4660.
- Renzi, P. and M. Bella, Non-asymmetric organocatalysis. Chemical Communications, 2012. 48(55): p. 6881-6896.
- Funatomi, T., et al., Pentafluorophenylammonium triflate (PFPAT): an efficient, practical, and cost-effective catalyst for esterification, thioesterification, transesterification, and macrolactone formation. Green Chemistry, 2006. 8(12): p. 1022-1027.
- Iida, A., et al., Mild and efficient pentafluorophenylammonium triflate (PFPAT)-catalyzed C-acylations of enol silyl ethers or ketene silyl (thio) acetals with acid chlorides. Organic letters, 2007. 9(10): p. 1859-1862.
- Khaksar, S. and S.M. Ostad, Pentafluorophenylammonium triflate as an efficient, environmentally friendly and novel organocatalyst for synthesis of bis-indolyl methane derivatives. Journal of Fluorine Chemistry, 2011. 132(11): p. 937-939.
- Ghashang, M., S.S. Mansoor, and K. Aswin, Pentafluorophenylammonium triflate (PFPAT) catalyzed facile construction of substituted chromeno [2, 3-d] pyrimidinone derivatives and their antimicrobial activity. Journal of advanced research, 2014. 5(2): p. 209-218.
- Montazeri, N., et al., Pentafluorophenylammonium triflate-CuCl 2: A mild, efficient and reusable heterogeneous catalyst system for facile synthesis of 4 (3H)-quinazolinones under solvent-free conditions. Journal of Chemical Sciences, 2012. 124(4): p. 883-887.
- Khaksar, S. and N. Behzadi, Pentafluorophenylammonium triflate (PFPAT): an efficient, practical, and cost-effective catalyst for one-pot condensation of β-naphthol, aldehydes and cyclic 1, 3-dicarbonyl compounds. Combinatorial chemistry & high throughput screening, 2012. 15(10): p. 845-848.
- Abdelraheem, E.M., et al., Two Step Macrocycle Synthesis by Classical Ugi Reaction. The Journal of organic chemistry, 2018.
- Khaksar, S. and M. Gholami, An eco-benign and highly efficient access to dihydro-1H-indeno [1, 2-b] pyridines in 2, 2, 2-trifluoroethanol. Journal of Molecular Liquids, 2014. 196: p. 159-162.
- Khaksar, S. and H. Radpeyma, Pentafluorophenylammonium triflate: A highly efficient catalyst for the synthesis of quinoxaline derivatives in water. Comptes Rendus Chimie, 2014. 17(10): p. 1023-1027.
- Khaksar, S. and S.M. Talesh, Three-component one-pot synthesis of 2, 3-dihydroquinazolin-4 (1H)-one derivatives in 2, 2, 2-trifluoroethanol. Comptes Rendus Chimie, 2012. 15(9): p. 779-783.
- Ram, V.J., et al., Oxygenated chalcones and bischalcones as potential antimalarial agents. Bioorganic & medicinal chemistry letters, 2000. 10(19): p. 2159-2161.
- Poupelin, J.P., et al., synthesis and anti-inflammatory properties of bis (2‐hydroxy‐1‐naphthyl) methane derivatives. I. Monosubstituted derivatives. ChemInform, 1978. 9(25).
- Hideo, T. and J. Teruomi, Jpn. Patent 56005480. Sankyo Co, 1981.
- Zelefack, F., et al., Cytotoxic and antiplasmodial xanthones from Pentadesma butyracea. Journal of natural products, 2009. 72(5): p. 954-957.
- Nguyen, H.T., et al., Antitumor psoropermum xanthones and sarcomelicope acridones: privileged structures implied in DNA alkylation. Journal of natural products, 2009. 72(3): p. 527-539.
- Hilderbrand, S.A. and R. Weissleder, One-pot synthesis of new symmetric and asymmetric xanthene dyes. Tetrahedron letters, 2007. 48(25): p. 4383-4385.
- Knight C. G., and Stephens T. (1989) Xanthene-dye-labelled phosphatidylethanolamines as probes of interfacial pH. Biochem. J., 258, 683-687.
- Sheffler DJ, Conn PJ. (2008) Allosteric potentiators of metabotropic glutamate receptor subtype 1a differentially modulate independent signaling pathways in baby hamster kidney cells. Neuropharmacology 55:419–427.
- Baguley, B.C. and G.J. Finlay, Derivatives of amsacrine: determinants required for high activity against Lewis lung carcinoma. JNCI: Journal of the National Cancer Institute, 1988. 80(3): p. 195-199.
- Dabiri, M.; Baghbanzadeh, M.; Nikcheh, MS; Arzroomchilar, E. Eco-friendly and efficient one-pot synthesis of alkyl- or aryl-14H-dibenzo[a,j]xanthenes in water. Bioorg. Med. Chem … Bioorg. Med. Chem. 2007, 17, 621–623.
Recieved: 01-03-2023 | Web published: 17-05-2022 | Views 644
